Summary of Selected Projects
Click to expand each project's details.
Summary of Selected Projects
Gene Therapy for Heritable Dilated Cardiomyopathy
There are no details avalable for this project.
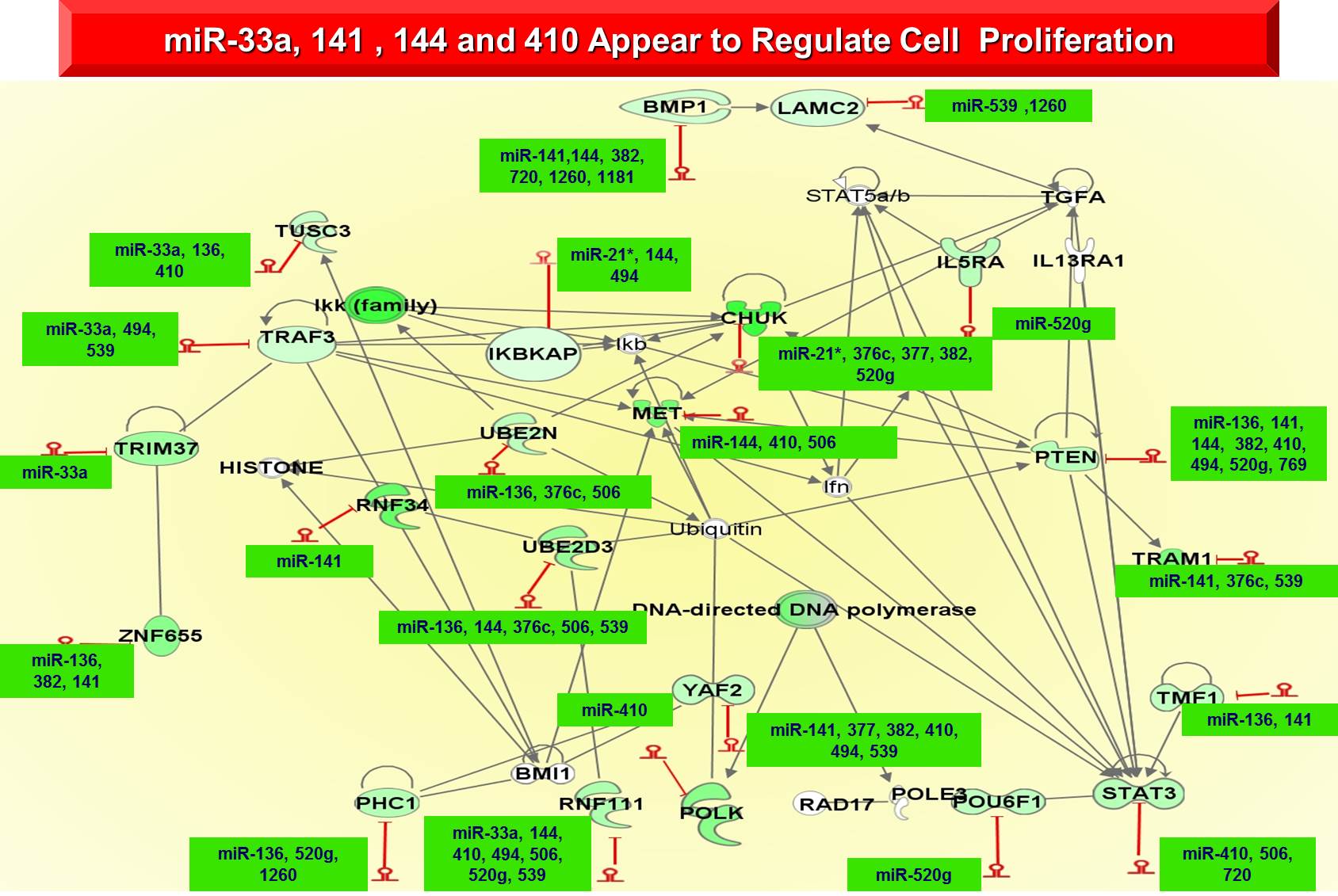
Role of a Novel Cardiac Glucose Transporter, SGLT1, in Heart Failure
There are two families of cellular glucose transporters, the facilitative glucose transporters (GLUTs) and the sodium-dependent glucose transporters (SGLTs). Although it was thought that only GLUT1 and GLUT4 are active in cardiomyocytes, we discovered that SGLT1 protein is present in cardiomyocytes. These findings were from our studies of a glycogen storage cardiomyopathy caused by gain-of-function mutations in the PRKAG2 gene that encodes the γ2 subunit of 5’-AMP-activated protein kinase (AMPK). To study cardiac SGLT1, we constructed two transgenic FVB strain mouse models: (1) cardiomyocyte-specific knockdown of SGLT1 (TGSGLT1-DOWN mice), using transgenic RNAi technology; and (2) cardiomyocyte-specific doxycycline-suppressible conditional overexpression of SGLT1 (TGSGLT1-ON mice). Using these models, we have shown that SGLT1 upregulation contributes to PRKAG2 glycogen storage cardiomyopathy. We next examined cardiac SGLT1 in diabetes and ischemia because AMPK is known to play a role in these conditions. We have found that pharmacological activation of AMPK upregulates SGLT1. TGSGLT1-DOWN mice are protected from cardiac injury following ischemia-reperfusion, and in type 1 and type 2 diabetes mellitus. The aims of our ongoing studies are:
- To determine the molecular and cellular mechanisms that regulate cardiac SGLT1 expression and activity in health and disease.
- To determine the effect of modulation of cardiac SGLT1 on ischemic heart failure and diabetic cardiomyopathy.
- To determine the molecular and cellular mechanisms by which SGLT1 contributes to ischemic heart failure and diabetic cardiomyopathy.
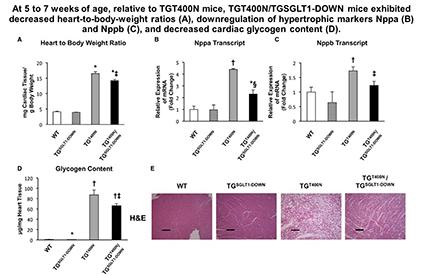
A Minipig Genetic Model for Hypertrophic Cardiomyopathy (HCM)
In collaboration with MyoKardia, we have generated Yucatan minipigs with a heterozygous knockin of the R403Q mutation in MYH7, a well-characterized human hypertrophic cardiomyopathy (HCM) mutation, using homologous recombination and somatic cell nuclear transfer. We have conducted deep phenotyping with biomechanical studies of myocardial tissue samples, circulating biomarker analysis, multi-modality cardiac imaging and histologic and multi-omic analysis of left ventricular tissue samples. R403Q pigs recapitulate the human phenotype, exhibiting an increase in left ventricular wall thickness by 9 months of age. The hypertrophy is preceded by early hypercontractility, subclinical ischemia and fibrosis.
The aims of our ongoing studies are:
- To determine whether epigenetic and regional hemodynamic differences underlie the heterogeneity of hypertrophy patterns (asymmetric vs concentric).
- To determine the role of protein misfolding and ER stress in HCM.
- To determine the electrophysiological abnormalities underlying HCM and sudden cardiac death using optical mapping and patch clamping.
- To determine the role of exercise in exacerbating or attenuating the HCM phenotype.
Accelerated Heart Failure in Heterozygous Carriers of Cystic Fibrosis Mutations
Cystic fibrosis (CF) results from mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated and ATP-gated chloride channel. CFTR is also expressed in cardiomyocytes and vascular smooth muscle cells, where its function is largely unknown. Despite the high prevalence of heterozygous CFTR variant carriers, little is known about how the heterozygous carrier state affects health. Primarily in animal studies, a functional role of CFTR has been implicated in myocardial hypertrophy and heart failure, cardiac ischemia/reperfusion injury, and vascular smooth muscle function. In collaboration with Dr. Barry London, we determined that heterozygosity for the most common CFTR variant (ΔF508) was associated with a higher incidence of death and the combined endpoint of death/ventricular assist device (VAD) placement/heart transplant in two large cohorts totaling ~2000 Caucasian subjects with heart failure. Therefore, CFTR variants may represent a heretofore unrecognized but common genetic modifier of heart failure outcomes. It is unknown whether decreased CFTR function worsens heart failure outcomes through direct effects on cardiomyocytes, such as perturbations of ion homeostasis and contractility, or through dysfunction in other organs such as exacerbation of lung edema. We hypothesize that heterozygous CFTR variant carriers have subclinical cardiac, pulmonary, and/or vascular dysfunction that predisposes them to the worse clinical outcomes. The aims of our ongoing studies are:
- To ascertain the effects of CFTR haploinsufficiency on cardiac and pulmonary function at baseline and following myocardial infarction (a heart failure model) in CFTR+/- and CFTR+/+ pigs, at the level of cells, organs, and intact pigs.
- To determine whether human heterozygous CFTR variant carriers exhibit subclinical cardiac, pulmonary, or vascular dysfunction.
- To determine the risk of developing heart failure and other cardiopulmonary outcomes associated with the CFTR variant carrier state in large medical insurance claims databases.
Dissecting Genetic Mechanisms of Hypertrophic Cardiomyopathy by ENU Mutagenesis
Previous human genetic studies have identified the etiology of many heritable cardiovascular disorders. Hypertrophic cardiomyopathy (HCM), the most common heritable cardiovascular disorder (prevalence 1:500), has been associated with mutations in at least 19 genes, most of which encode sarcomere proteins, along with a few encoding Z-disc and calcium handling proteins. However, mutations are detected in previously identified genes in only ~50% of patients. Therefore, we hypothesize that variants in other heretofore undiscovered genes that regulate the sarcomere, the Z-disc, or cellular calcium homeostasis, including metabolic genes, can directly cause disease. Large scale forward genetics in mice is a novel approach to identifying these undiscovered genes. The NHLBI has funded a mouse ethylnitrosourea (ENU) mutagenesis program that is identifying genetic pathways contributing to congenital heart disease. Preliminary studies show that 6% of fetuses homozygous for a given mutation have isolated cardiac hypertrophy. Therefore, our overall objective is to leverage the unique resource available to us through this ENU mutagenesis program to recover novel genes associated with HCM. Our aims of our ongoing studies are:
- To recover ENU mutagenized mouse pedigrees with HCM.
- To identify novel genes with mutations leading to HCM.
Identification of Novel Genetic and Genomic Mechanisms of Cardiomyopathies in Human Subjects
The long-term response of the heart to a wide range of injuries is maladaptive ventricular growth and remodeling characterized by dilation and/or hypertrophy. This process ultimately leads to heart failure, which is associated with a 20% lifetime risk and is the most common cause of death in the United States. The long-term objective of our research program is to elucidate the molecular basis for ventricular remodeling, ultimately uncovering therapeutic targets. Cardiomyopathies are primary disorders of cardiac muscle (myocardium) characterized by cardiac hypertrophy or dilation, and associated with heart failure, arrhythmias, and sudden cardiac death. These disorders result from heritable genetic mutations, and thus represent genetic models of cardiac hypertrophy and dilation. Using high-throughput next generation sequencing technologies, we are screening a cohort of approximately 2000 unrelated human subjects with cardiomyopathies for pathogenic variants in genes not previously associated with heart disease. In addition, we are identifying modifier variants that exacerbate or attenuate the phenotype.
Gene Networks Beyond Organ Boundaries: Heart, Lung and Pulmonary Vascular Disease
hronic lung, heart and vascular diseases such as idiopathic pulmonary fibrosis (IPF), idiopathic pulmonary arterial hypertension (IPAH) secondary pulmonary hypertension (PH), and left ventricular (LV) and right ventricular (RV) heart failure affect a significant portion of the population and are collectively the most common causes of mortality and morbidity. Although the molecular mechanisms of individual diseases have been extensively studied over the last decades, it is unknown whether the separate organ diseases that sometimes share risk factors and co-occur in the same patient also share similar molecular mechanisms. Our overall objective is to use high throughput approaches and the extensive resources of well-characterized tissues to identify molecular phenotypes that are common or distinct across and within disease and organ boundaries. To identify such phenotypes we will:
- Identify tissue molecular signatures that are common in diseased tissue in IPF, IPAH, secondary PH, and RV and LV heart failure by analyzing failing and non-failing human RV tissue in IPAH or secondary PH; failing and non-failing human LV tissue; lung tissue from human subjects with IPF and control lungs; lungs and pulmonary vessels of patients with IPAH and secondary PH; and lung, RV, LV and pulmonary vessels from the same patient from our unique warm autopsy program. We will perform mRNA and microRNA expression profiling on the samples, validate key patterns and pathways by high throughput qRT PCR and generate cross organ tissue microarrays to perform high-throughput tissue protein validation and localization.
- Identify biomarkers of disease presence, stage and outcome, within and across organ and disease boundaries, in easily accessible peripheral blood by analyzing peripheral blood from patients with LV and RV failure, IPAH and IPF. We will perform mRNA and microRNA expression profiles and determine expression patterns that predict disease presence, state and outcome within and across organ and disease boundaries.
- Generate a disease and mechanism relevant transcriptional map in RV and LV failure, IPAH and IPF by performing an integrated analysis of mRNA and microRNA expression patterns as well as clinical data with the use of advanced computational approaches, followed by cell culture and animal model validations of analytic predictions.